1. De meest relevante punten
- Geen ijzersuppletie voorschrijven bij anemie bij personen met migratieachtergrond zonder analyse voor Hb-pathie.
- Denk ook aan Hb-pathie bij patiënten met voorouders met een migratieachtergrond.
- Homozygote vorm sikkelcelziekte (HbSS) en beta thalassemie major zijn ernstige ziektebeelden.
- Belangrijk: vaststellen, snel en adequaat behandelen en voorkomen provocerende factoren voor sikkelcelcrise: koorts, infectie, dehydratie, pijn, koude, stress, hoogte.
In Nederland is ± 1,1% van de bevolking drager van een hemoglobineafwijking. Dit percentage is hoger bij personen met een migratieachtergrond:
- Mensen met een migratieachtergrond en/of ouders uit Afrika, Suriname of Caribisch gebied: prevalentie dragerschap 10 – 24%.
- Mensen met een migratieachtergrond en/of ouders uit China/Oost-Aziatische landen: prevalentie dragerschap 3 – 26%.
- Mensen met een migratieachtergrond en/of ouders uit Middellandse Zee gebied, Turkije, Marokko: prevalentie dragerschap 0,5 – 8%.
Naar schatting leven in Nederland 2500 – 3000 patiënten met sikkelcelziekte.
In Nederland neonatale screening (hielprik) sinds 2007:
- per jaar 35 – 40 kinderen met sikkelcelziekte (HbSS) en 5 kinderen met bèta-thalassemie major gevonden
- per jaar 800 dragers van sikkelcelziekte gevonden
Per jaar worden ook ongeveer 60 nieuwe diagnoses bij niet-pasgeborenen gesteld.
Geografische distributie:
- Alpha-thalassemie: vooral in Zuid-Oost-Azië en China.
- Bèta-thalassemie major: rond Middellandse Zee (vooral Italië, Griekenland en Turkije), Midden-Oosten, Zuid-Oost-Azië en enkele gebieden in Afrika.
- Sikkelcelziekte: Afrika, Caribisch gebied, Mediterrane gebied, Midden-Oosten en Azië.
- Sikkelcelziekte: structuurdefect globineketens, autosomaal recessief.
- Thalassemieën: expressiedefect globineketens (afwezige of verminderde productie), autosomaal recessief.
- Er zijn dragers (heterozygoot) en patiënten (homozygoot). Twee dragers vormen samen een risicopaar dat per zwangerschap 25% kans heeft op een kind met de ernstige afwijking.
- Er komen veel combinaties voor van alpha/bèta-thalassemie en sikkelcelziekte, bijvoorbeeld circa 30% van de mensen met sikkelcel-hemoglobine heeft ook een alpha-thalassemie.
- Alpha-thalassemie:
- Over het algemeen milde asymptomatische microcytaire anemie.
- Bij deletie van 3 alpha-globine genen: klinische presentatie variërend van milde microcytaire anemie tot ernstig beeld met transfusie afhankelijkheid.
- Als alle 4 alpha-globine genen zijn uitgeschakeld: ernstig ziektebeeld met hydrops foetalis en overlijden voor of rondom geboorte.
- Bèta-thalassemie:
Bèta-thalassemie minor (dragerschap):
-
- Lichte microcytaire anemie, in principe geen klachten.
Bèta-thalassemie intermedia = tussenvorm:
-
- Zeer wisselend beloop, waarbij kliniek afhankelijk van de nog aanwezige hoeveelheid bèta-globine lijkt op presentatie van minor of major variant.
Bèta-thalassemie major (homozygoot):
-
- Progressieve, zeer ernstige anemie.
- Bleek-geel zien, kortademigheid, slechte groei; op den duur botvervormingen (‘chipmunk face’) en hepatosplenomegalie ten gevolge van extramedullaire hematopoiese.
- Bloedtransfusie afhankelijk.
- IJzerstapeling door vele transfusies.
- Sikkelcel-bèta-thalassemie
- Een combinatie van bèta-thalassemie en sikkelcelziekte.
- Minder ernstig dan sikkelcelziekte maar bloedtransfusies wel vaak nodig.
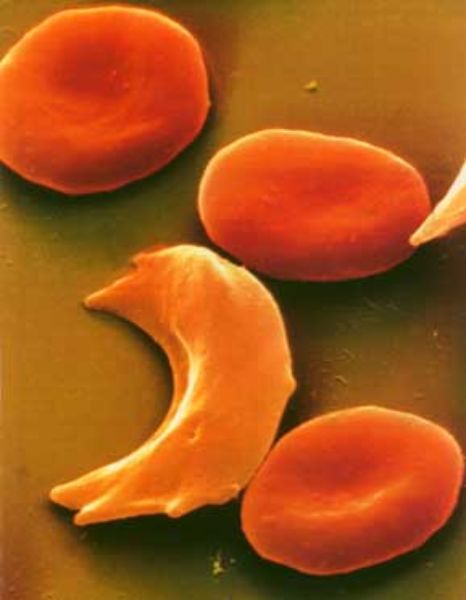
- Sikkelcelziekte:
- Sikkelcel dragerschap (HbS) geeft geen ziekteverschijnselen in het dagelijkse leven, maar potentieel wel onder extreme omstandigheden (zoals mariniers-training, verblijf in hooggebergte, etc).
- Homozygote sikkelcelziekte (HbSS) is een ernstige aandoening, alhoewel mate van ernst kan wisselen.
- Twee pathofysiologische mechanismen: afsluiten bloedvaten door sikkelcellen (vaso-occlusieve sikkelcelcrise) en kortere levensduur erythrocyt waardoor chronische hemolytische anemie en risico op anemische crise.
- Daarnaast verhoogde kwetsbaarheid voor infecties met gekapselde bacteriën door slechte tot afwezige milt-functie.
- Baby ontwikkelt niet direct symptomen, wordt eerste 3 – 4 maanden nog beschermd door foetaal hemoglobine (HbF). Eerste symptomen kunnen pijnlijke en gezwollen handen en/of voeten, anemie/icterus of ernstige infecties zijn.
- Levensverwachting gemiddeld 40 – 50 jaar
- Acute symptomen: sikkelcelcrise:
- kan variëren in aard en intensiteit: patiënt weet zelf vaak het beste of er sprake is van een crise
- uitlokkende factoren zijn infectie, warmte of koude, hypoxie, stress, dehydratie, maar vaak ook spontaan
- vaso-occlusieve crise: acute heftige pijnaanvallen in bot, spier, buik of penis (priapisme)
- anemische crise: aplastisch (door bijvoorbeeld Parvo B19 infectie), hyperhemolyse of acute milt- of leversequestratie
- ‘acute chest syndrome’ kenmerkt zich door respiratoire symptomen als dyspnoe of hoesten, koorts, thoracale pijn en/of extra zuurstofbehoefte met een nieuw infiltraat op X-thorax
- Chronische complicaties:
- chronische gewrichtspijnen
- achterblijvende groei
- verhoogd risico op ernstige infecties
- botvervormingen ten gevolge van extramedullaire hematopoiese
- neurologische complicaties (o.a. CVA)
- nierfalen
- retinopathie
- been ulcera
- galstenen
- Denk aan Hb-pathie bij personen met (voorouders met) een migratieachtergrond met anemie, icterus, acute pijn ledematen, rug of buik of ernstig verlopende pneumonie/ meningitis.
- Diagnostiek
- Volledig bloedbeeld, erythrocyten indices, reticulocyten (microcytair bloedbeeld, anemie).
- HPLC (High Performance Liquid Chromatography) voor sikkelcelziekte en beta thalassemie major, DNA diagnostiek voor alpha thalassemie.
- Beleid bij patiënten zonder klachten
Naar aanleiding van hielprik: handel conform RIVM draaiboek hielprikscreening:- bij melding van vermoeden sikkelcelziekte of bèta-thalassemie major:
- informeer ouders:
- uitslag moet nog bevestigd worden
- als uitslag juist, symptomen niet voor leeftijd van 3-4 maanden
- voorzorgmaatregelen zijn nodig om ziekteverschijnselen op termijn te voorkomen/ beperken
- geef ouders indien gewenst een informatieblad van het RIVM (Zie links voor patiënten onder 6.)
- verwijs baby naar kinderhematoloog academisch ziekenhuis
- verwijs ouders naar Kl. Genetisch centrum
- informeer ouders:
- bij melding van dragerschap:
- informeer ouders:
- dragerschap is geen ziekte
- belangrijk om te weten of beide ouders drager zijn
- indien beide ouders drager: vlokkentest bij evt. toekomstige zwangerschap mogelijk bij 10e zwangerschapsweek
- geef ouders indien gewenst een informatieblad van het RIVM (Zie links voor patiënten onder 6.)
- bied beide ouders onderzoek naar dragerschap aan
- indien één ouder drager, geen risico; beide ouders drager à Genetisch centrum
- informeer ouders:
- verwijzing zwangeren met migrantenachtergrond zo vroeg mogelijk in zwangerschap voor labscreening. Indien drager: screening partner; indien partner positief is prenatale diagnostiek mogelijk rond 10e week zwangerschap.
- overweeg het bespreken van dragerschapstest bij zwangerschapswens.
- bij melding van vermoeden sikkelcelziekte of bèta-thalassemie major:
- Beleid bij patiënten met klachten:
Afhankelijk van leeftijd, ernst en symptomen en complicaties; verwijzen naar/in overleg met (kinder)hematoloog:- ernstige thalassemie:
- erythrocyten transfusies
- therapeutische interventies om ijzerstapeling zoveel mogelijk te voorkomen
- stamcel transplantatie voorlopig enige curatieve therapie
- sikkelcelziekte
- penicillineprofylaxe tot de leeftijd van 12 jaar
- foliumzuur suppletie
- extra vaccinaties
- eventueel hydroxycarbamide bij frequente crises
- erythrocyten transfusies
- tijdens crises: vocht en analgetica
- ernstige thalassemie:
- NHG-Standaard Anemie, versie 2.1, Nederlands Huisartsen Genootschap Oktober 2014.
- Hemoglobinopathie in Nederland: een exoot op klompen. Ned Tijdschr Hematol 2014;11:44-54
- Voor professionals:
- oscarnederland.nl: patiëntenorganisatie, informatie voor professionals en leken
- HbPinfo.com voor professionals en leken, folders in meerdere talen
- Richtlijn behandeling sikkelcelziekte
- Werkboek kinderhematologie, thalassemie
- Draaiboek hielprikscreening
- Hemoglobinopathieen
- Hemoglobinopathie in Nederland: een exoot op klompen. Ned Tijdschr Hematol 2014;11:44-54
- Kolnaar B & Grol M. De rol van de huisarts bij hemoglobinopathie H&W 46 (2003): 340-341
- Giordano PC, Poland D, Harteveld CL. Neonatale screening voor hemoglobinopathie. H&W 54(6) juni 2011:
- Giordano PC & Harteveld CL. Prevention of hereditary haemoglobinopathies in the Netherlands. Tijdschr. Geneeskd 2006 sep 30; 150 (39) 2137-41. Review, Dutch
- Voor patienten:
- oscarnederland.nl: patiëntenorganisatie
- HbPinfo.com folders in meerdere talen
- Thuisarts sikkelcelziekte
- Cyberpoli sikkelcelziekte
- Thuisarts thalassemie
- Cyberpoli thalassemie
- RIVM hielprikscreening informatieblad alphathalassemie
- RIVM hielprikscreening informatieblad beta-thalassemie
- RIVM hielprikscreening informatieblad sikkelcelziekte
- RIVM folder ‘Uw kind is drager van sikkelcel’
- Info over dragerschapsonderzoek (ook preconceptioneel) bij mensen afkomstig uit bepaalde streken.
- Info over dragerschapsonderzoek erfelijke bloedarmoede
anemie, hb-pathie, hemoglobinopathie, sikkelcel, sikkelcelziekte, thalassemie, sikkelcelcrise, crise.
